nf-core/variantprioritization
Analysis pipeline for the functional annotation and translation of somatic SNVs/InDels and copy number aberations for precision cancer medicine.
Introduction
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
- Reference data – PCGR and VEP resources when downloaded or provided as archives
- VCF preprocessing – optional bgzip/tabix, left-normalisation and filtering
- Variant formatting and intersection – reheader VCFs, merge caller support, and reformat CNAs
- PCGR – combined somatic variant annotation and reporting
- CPSR – combined germline variant annotation and reporting
- MultiQC – aggregate QC and provenance reporting
- Pipeline information – run metadata and software versions
Reference data
Output files
reference/- PCGR reference bundle extracted from
--pcgr_downloador a provided--pcgr_database_dirtarball. - VEP cache extracted from a provided archive when supplied via
--vep_cacheas a.tar.gzbundle.
- PCGR reference bundle extracted from
Reference resources are only written to the results directory when you request downloads (--pcgr_download) or provide archived bundles. When pointing to pre-existing directories, the pipeline reuses them without copying.
VCF preprocessing
Output files
tabix/*.tbi: tabix index files created for input VCFs that were missing an index (for exampletabix/HCC1395T_vs_HCC1395N.mutect2.filtered.vcf.gz.tbi).
bcftools/norm/{sample}.{caller}.norm.vcf.gzand.tbi: left-aligned, normalized VCFs produced bybcftools normusing the reference FASTA.
bcftools/filter/{sample}.{caller}.norm.filtered.vcf.gzand.tbi: filtered VCFs (using thebcftools filtermodule expression configured inmodules.config) retaining indexed output.
Input VCFs are optionally bgzipped and indexed, then normalized and filtered per caller (e.g. Mutect2, Strelka). Output paths retain caller-specific prefixes to make downstream merging transparent.
Variant formatting and intersection
Output files
custom/intersect_vcf/{sample}_keys.txt: table mapping each variant to the set of callers that support it (column 5 lists caller names). Used to propagate caller provenance into downstream VCFs.
custom/reformat/{sample}.{caller}.reformatted.vcf.gzand.tbi: caller VCFs rewritten byreformat_vcf.pyto add INFO tags such asTDP,NAF,TAF,ADT,ADN, and caller codes (TAL/AL). Tumor/normal order is auto-detected and the header is updated accordingly.{sample}.reformatted.allelic_cna.tsv: allele-specific CNA table created from CNVkit or ASCAT input viareformat_cna.py(Chromosome,Start,End,nMajor,nMinor).
custom/pcgr_ready_vcf/{sample}.vcf.gzand.tbi: unified somatic VCF built by combining reformatted caller VCFs and the{sample}_keys.txtcaller map. This file is ready for PCGR.
Variants are first intersected across somatic callers to capture support per tool. VCFs are then harmonised (depth/AF fields, caller codes) and merged into a single PCGR-ready VCF per sample. CNAs are reformatted into the allele-specific schema expected by PCGR when CNA analysis is enabled.
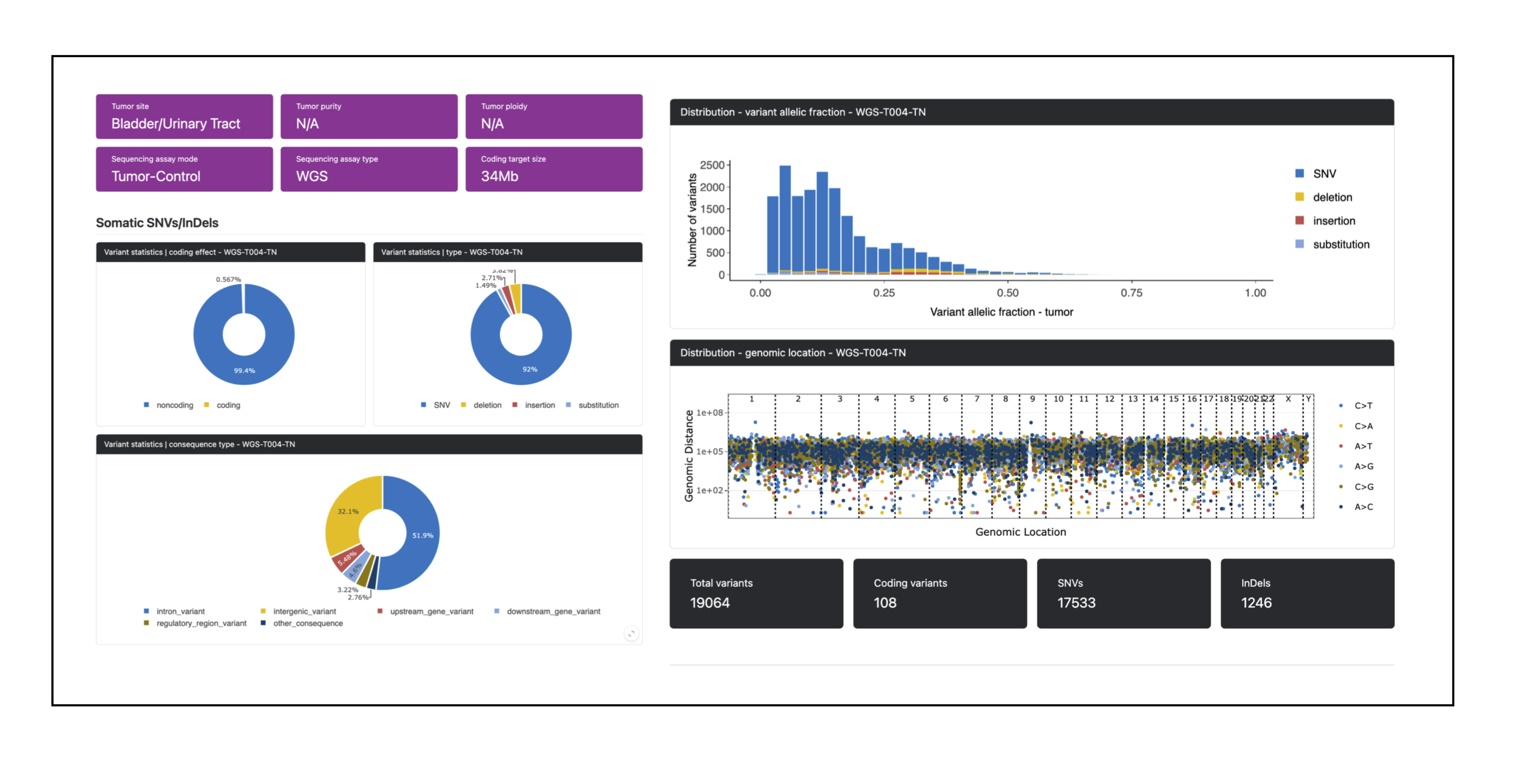
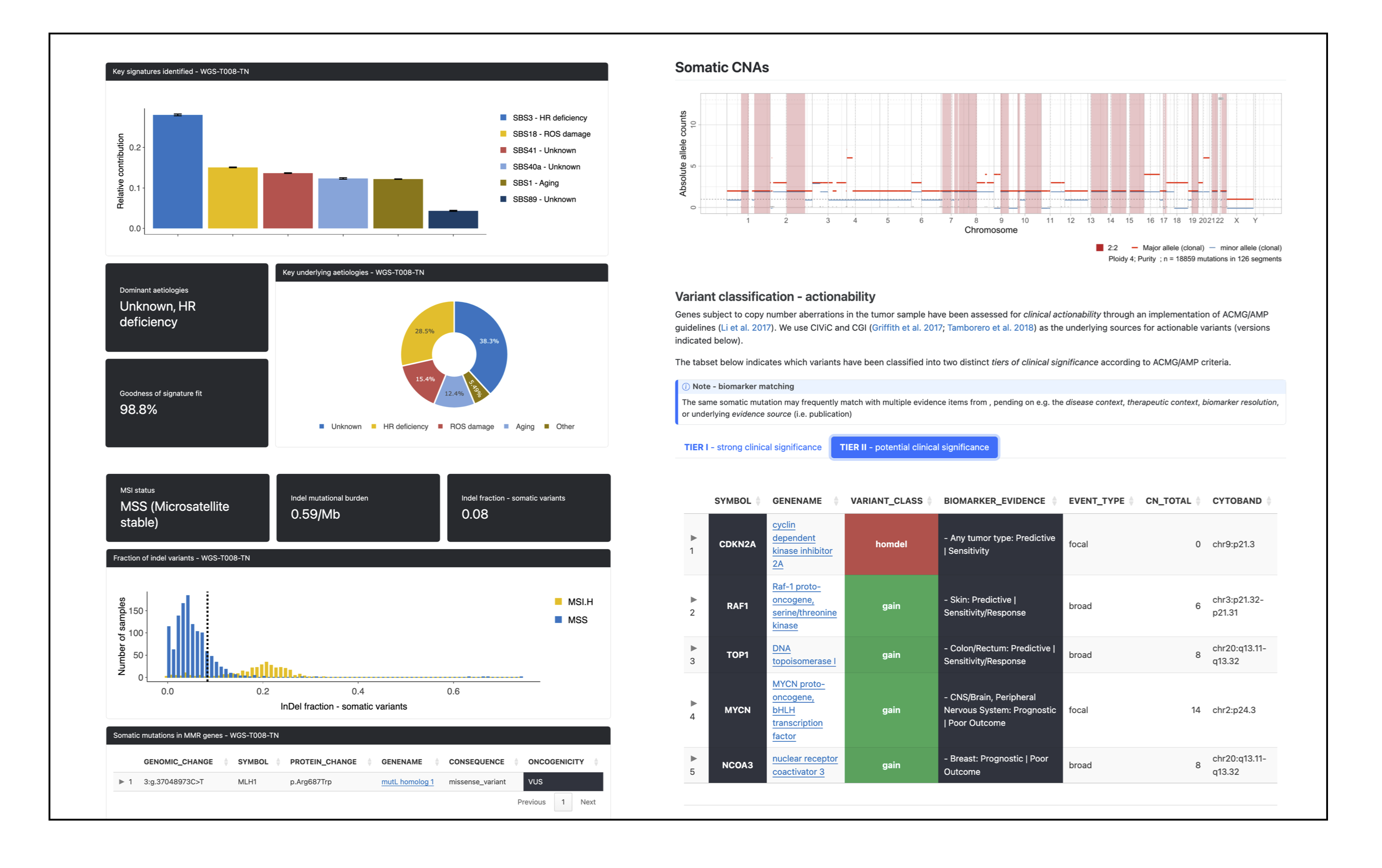
PCGR
Output files
pcgr/{sample}/{sample}.pcgr.{assembly}.html: interactive PCGR report.{sample}.pcgr.{assembly}.xlsx: Excel summary of annotated variants.{sample}.pcgr.{assembly}.maf: MAF-formatted somatic variants.{sample}.pcgr.{assembly}.pass.*: PASS-filtered TSV/VCF summaries (e.g..pass.tsv.gz,.pass.vcf.gz+.tbi).{sample}.pcgr.{assembly}.snv_indel_ann.tsv.gz,msigs.tsv.gz,tmb.tsv,cna_gene*.tsv.gz,cna_segment.tsv.gz: auxiliary annotation tables.{sample}.pcgr.{assembly}.conf.yaml: configuration used by PCGR.
PCGR ingests the unified somatic VCF (and optional allele-specific CNA table) together with the provided reference bundle and VEP cache to produce interactive and machine-readable reports per sample.
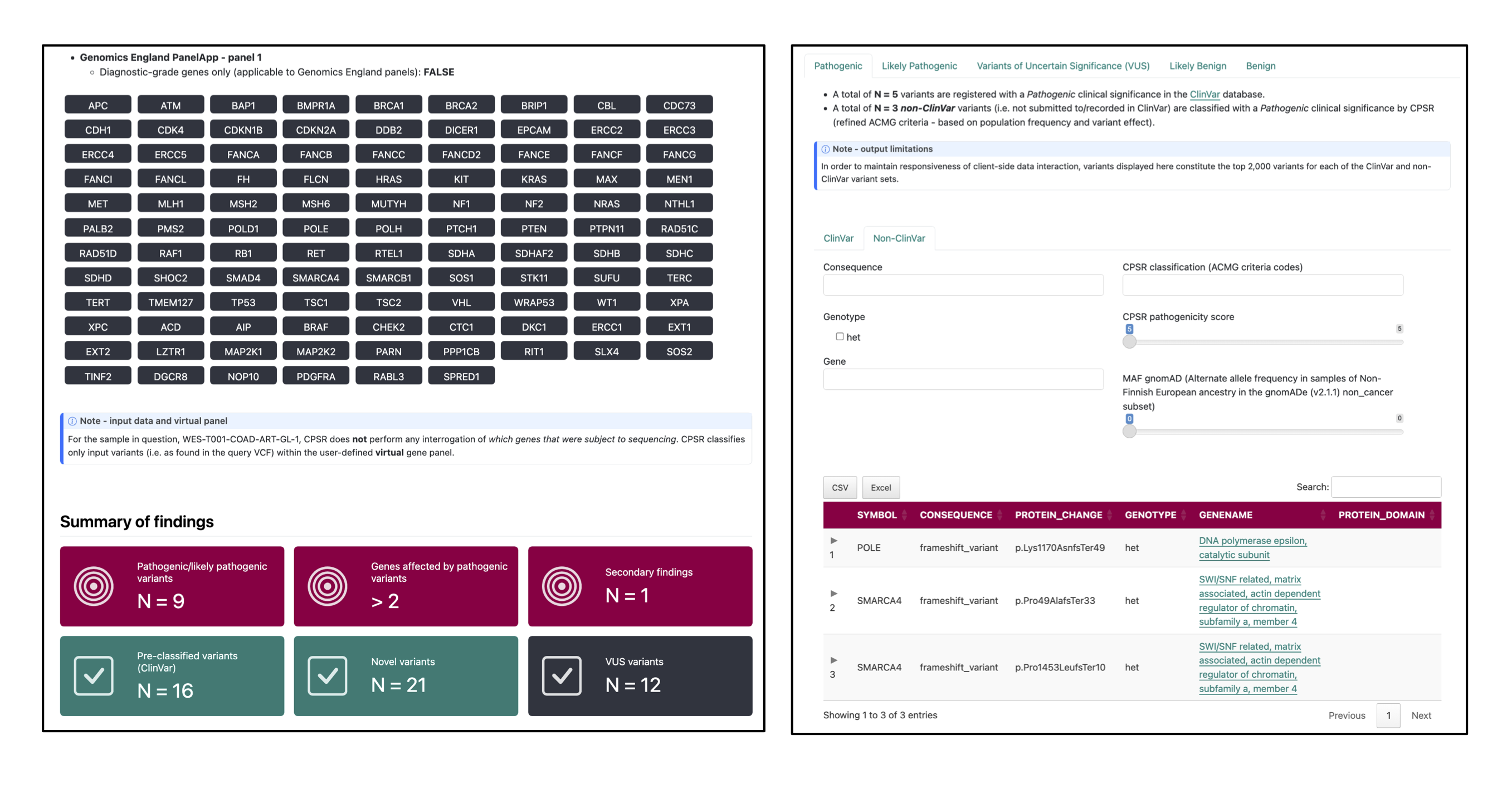
CPSR
Output files
cpsr/{sample}/{sample}.cpsr.{assembly}.html: interactive CPSR report.{sample}.cpsr.{assembly}.xlsx: Excel summary of annotated variants.{sample}.cpsr.{assembly}.pass.*: PASS-filtered TSV/VCF summaries (e.g..pass.tsv.gz,.pass.vcf.gz+.tbi).{sample}.cpsr.{assembly}.conf.yaml: configuration used by CPSR.
CPSR ingests each germline VCF together with the provided reference bundle and VEP cache to produce interactive and machine-readable reports per sample.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: directory containing static images from the report in various formats.
MultiQC is a visualization tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in the report data directory.
Results generated by MultiQC collate pipeline QC from supported tools e.g. FastQC. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter’s are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv. - Parameters used by the pipeline run:
params.json.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.
Exemplary Output
PCGR

CPSR