nf-core/sopa
Nextflow version of Sopa - spatial omics pipeline and analysis
Introduction
nf-core/sopa is the Nextflow version of Sopa. Built on top of SpatialData, Sopa enables processing and analyses of spatial omics data with single-cell resolution (spatial transcriptomics or multiplex imaging data) using a standard data structure and output. We currently support the following technologies: Xenium, Visium HD, MERSCOPE, CosMX, PhenoCycler, MACSima, Molecural Cartography, and others. It outputs a .zarr directory containing a processed SpatialData object, and a .explorer directory for visualization.
If you are interested in the main Sopa python package, refer to this Sopa repository. Else, if you want to use Nextflow, you are in the good place.
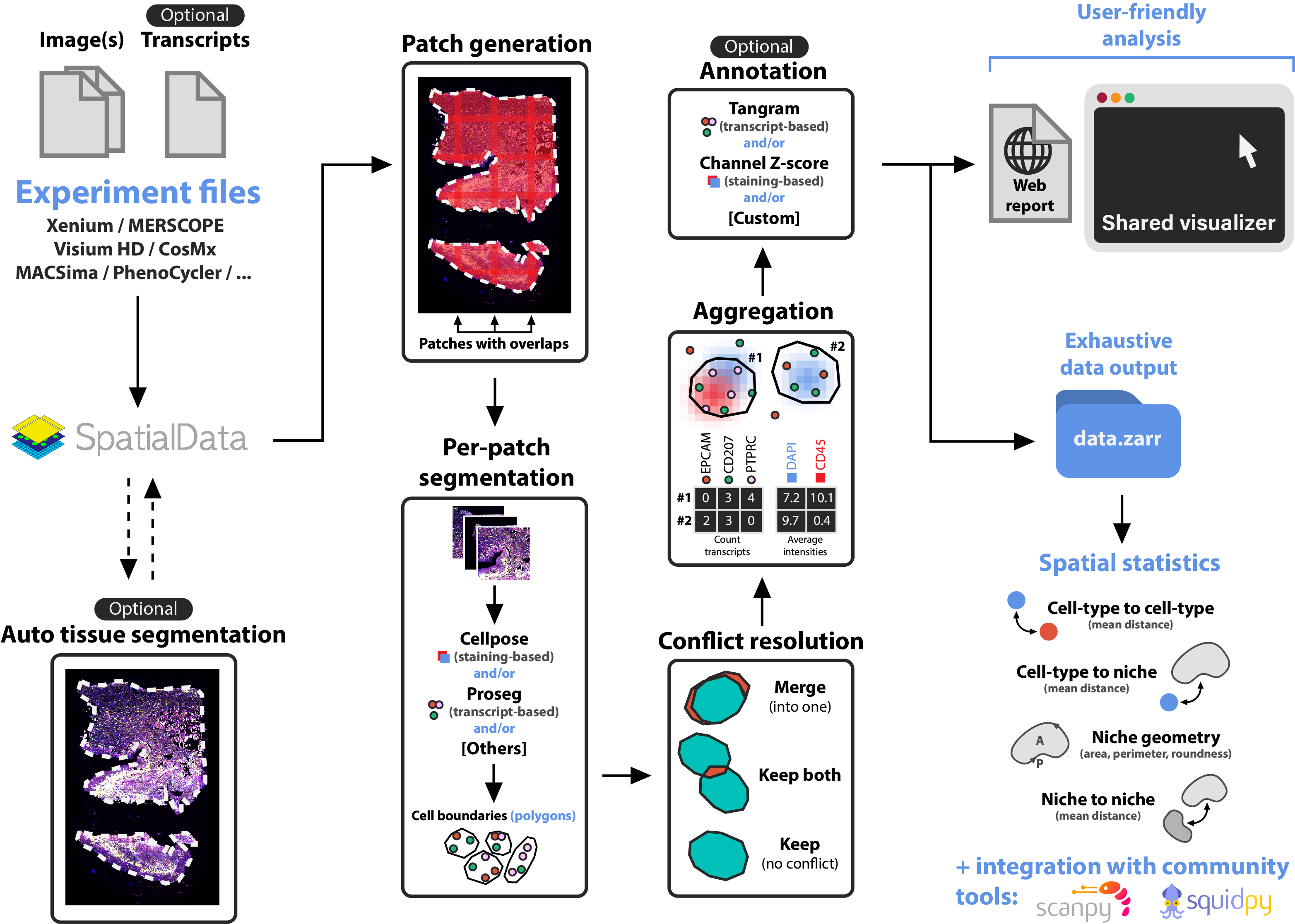
- (Visium HD only) Raw data processing with Space Ranger
- (Optional) Tissue segmentation
- Cell segmentation with Cellpose, Baysor, Proseg, Comseg, Stardist, …
- Aggregation, i.e. counting the transcripts inside the cells and/or averaging the channel intensities inside cells
- (Optional) Cell-type annotation
- User-friendly output creation for visualization and quick analysis
- Full SpatialData object export as a
.zarrdirectory
After running nf-core/sopa, you can continue analyzing your SpatialData object with sopa as a Python package.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet that lists the data_path to each sample data directory (typically, the per-sample output of the Xenium/MERSCOPE/etc, see more info here). You can optionally add sample to provide a name to your output directory, else it will be named based on data_path. Here is a samplesheet example:
samplesheet.csv:
sample,data_pathSAMPLE1,/path/to/one/merscope_directorySAMPLE2,/path/to/one/merscope_directoryIf you have Visium HD data, the samplesheet will have a different format than the one above. Directly refer to the usage documentation and the parameter documentation.
Then, choose the Sopa parameters (denoted below as <TECHNOLOGY_PROFILE>). To do that, you can provide an existing -profile containing all the dedicated Sopa parameters, depending on your technology, see the available technology-specific profiles here.
Now, you can run the pipeline using:
nextflow run nf-core/sopa \ -profile <docker/singularity/.../institute>,<TECHNOLOGY_PROFILE> \ --input samplesheet.csv \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/sopa was originally written by Quentin Blampey during his work at the following institutions: CentraleSupélec, Gustave Roussy Institute, Université Paris-Saclay, and Cure51.
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #sopa channel (you can join with this invite).
Citations
If you use nf-core/sopa for your analysis, please cite it using the following doi: 10.5281/zenodo.21382875
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the sopa publication as follows:
Sopa: a technology-invariant pipeline for analyses of image-based spatial omics.
Quentin Blampey, Kevin Mulder, Margaux Gardet, Stergios Christodoulidis, Charles-Antoine Dutertre, Fabrice André, Florent Ginhoux & Paul-Henry Cournède.
Nat Commun. 2024 June 11. doi: 10.1038/s41467-024-48981-z
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.