nf-core/longraredisease
Long read sequencing pipeline to identify variants in patients with neurodevelopmental disorders
Introduction
nf-core/longraredisease is a specialized bioinformatics pipeline for structural variant (SV) detection and clinical interpretation from long-read sequencing data (Oxford Nanopore and PacBio). Designed for rare disease diagnostics, it delivers high-confidence variant discovery through multi-caller consensus, family-based analysis, and phenotype-driven prioritization.
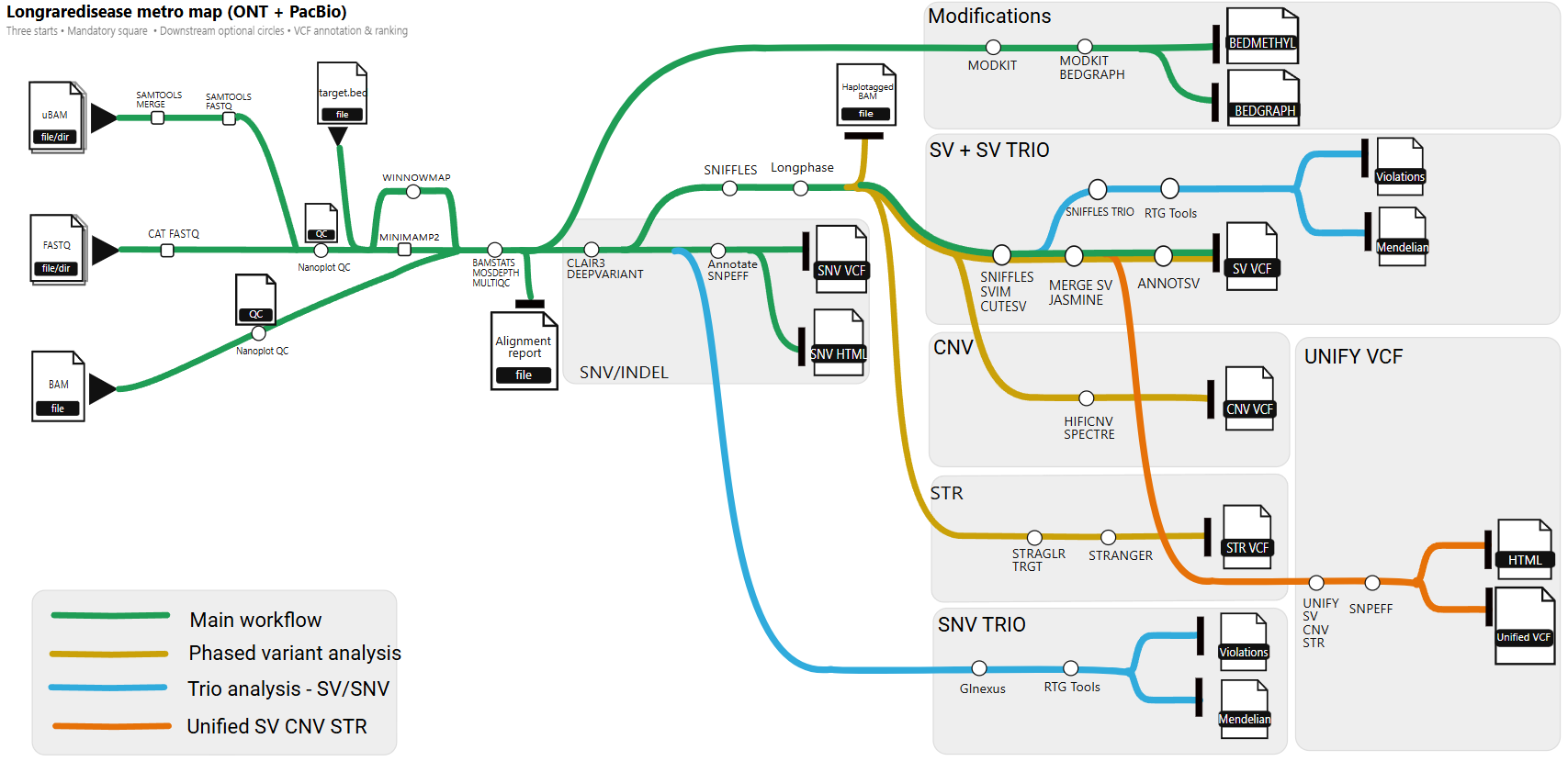
The pipeline supports:
- Multi-caller SV consensus — Sniffles, CuteSV, SVIM with JASMINE merging
- Phase-aware calling — Haplotype-resolved SV detection using LongPhase
- Family analysis — Trio-based joint calling and de novo variant detection
- Clinical annotation — AnnotSV with disease database integration
- Phenotype prioritization — SVANNA-based ranking using HPO terms
- Optional analyses — SNVs (Clair3/DeepVariant), CNVs (Spectre/HiFiCNV), STRs (Straglr), Methylation (Modkit)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data:
sample,file_path,hpo_terms,sex,phenotype,family_id,maternal_id,paternal_idsample1,/path/to/sample1.bam,HP:0002721;HP:0002110,1,2,,,Now, you can run the pipeline using:
nextflow run nf-core/longraredisease \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR> \ --fasta reference.fasta \ --sequencing_platform ontPlease provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided with the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details on pipeline usage and parameters, see docs/usage.md.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to docs/output.md.
Credits
I thank the following people for their contributions and guidance to the development of the pipeline:
The nf-core team, and especially Friederike Hanssen, Ken Brewer, Nicolas Vannieuwkerke and Maxime U Garcia for their support and guidance in developing this pipeline.
I also thank the clinical scientists Chipo Mashayamombe-Wolfgarten, Hannah Titheradge, and Lorraine Hartles-Spencer for their invaluable clinical input and expertise. I would also like to thank Professor Andrew Beggs for his clinical guidance and expertise.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #longraredisease channel (you can join with this invite).
Citations
If you use nf-core/longraredisease for your analysis, please cite it using the following doi: 10.5281/zenodo.20935122
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.