nf-core/pacsomatic
Nextflow pipeline for PacBio HiFi tumor/normal somatic genomics
Introduction
nf-core/pacsomatic is a bioinformatics best-practice pipeline for somatic variant analysis using PacBio HiFi sequencing data from matched tumor-normal samples.
The pipeline performs comprehensive somatic analysis including:
- Variant calling: SNVs, indels, structural variants (SVs), and copy number variants (CNVs)
- Variant annotation: Functional annotation and mutation signatures
- Methylation analysis: CpG methylation calling and differential methylation region (DMR) detection
- Tumor characterization: Clonality, purity, ploidy, and homologous recombination deficiency (HRD) analysis
The pipeline is built using Nextflow, a workflow manager to run tasks across multiple compute infrastructures in a portable, reproducible manner. It is designed following the nf-core community’s best practices and utilizes containerization with Docker, Singularity, or Conda for dependency management.
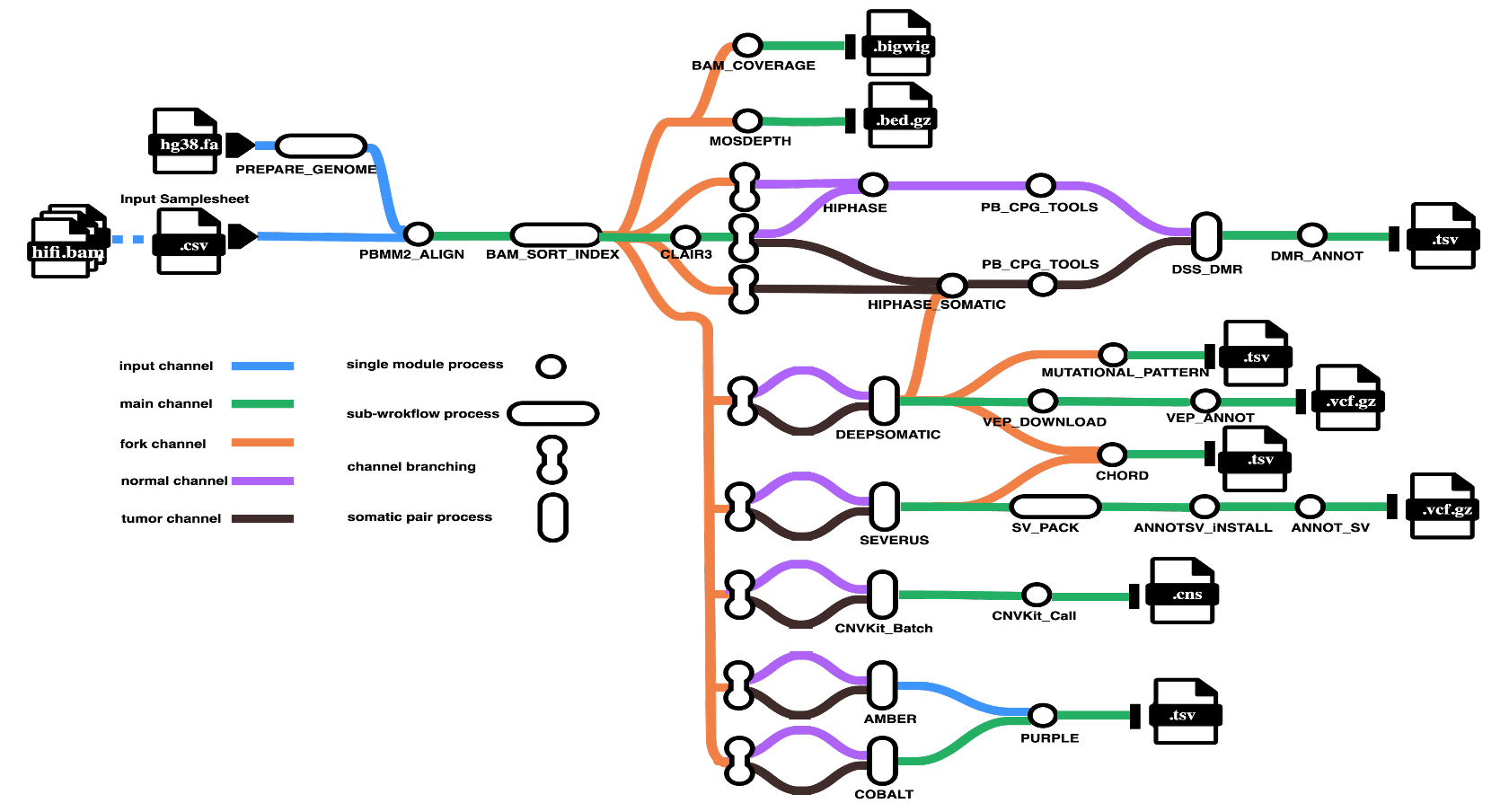
Pipeline Overview
The pipeline performs the following steps:
1. Alignment and Quality Control
- Read alignment to reference genome (pbmm2)
- Alignment sorting and indexing (SAMtools)
- Coverage analysis (bamCoverage, mosdepth)
- Aggregated quality reporting (MultiQC)
2. Variant Calling
- Germline SNVs: Clair3
- Variant phasing: HiPhase
- Somatic SNVs/indels: DeepSomatic
- Somatic structural variants: Severus
- Somatic copy number variants: CNVkit
3. Variant Annotation and Filtering
- SNV/indel functional annotation (VEP)
- Mutation signature analysis (MutationalPatterns)
- SV filtering (svpack)
- SV annotation (AnnotSV)
4. Methylation Analysis
- CpG methylation calling (pb-CpG-tools)
- Differential methylation region detection (DSS)
- DMR annotation (annotatr)
5. Tumor Characterization
- Homologous recombination deficiency estimation (CHORD)
- Tumor purity and ploidy analysis (AMBER, COBALT, PURPLE)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
Minimal samplesheet format (samplesheet.csv):
patient,sample,status,bamID1,S1_tumor,1,/path/to/ID1_S1_tumor.bamID1,S1_normal,0,/path/to/ID1_S1_normal.bamID2,S2_tumor,1,/path/to/ID2_S2_tumor.bamID2,S2_normal,0,/path/to/ID2_S2_normal.bamExtended samplesheet with PBI index files:
patient,sample,status,bam,pbiID1,S1_tumor,1,/path/to/ID1_S1_tumor.bam,/path/to/ID1_S1_tumor.bam.pbiID1,S1_normal,0,/path/to/ID1_S1_normal.bam,/path/to/ID1_S1_normal.bam.pbiColumn descriptions:
patient: Unique patient identifier (samples with the same ID are treated as matched pairs)sample: Unique sample identifierstatus: Sample type (1= tumor,0= normal)bam: Full path to unaligned BAM filepbi: (Optional) Full path to PacBio index (.pbi) file
Now, you can run the pipeline using:
nextflow run nf-core/pacsomatic \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR> \ --genome GRCh38Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline Output
Results are organized into functionally grouped subdirectories:
results/├── alignment/ # Aligned BAMs and QC metrics│ ├── pbmm2/ # Aligned BAM files│ └── qc/ # Alignment quality control├── germline_snv/ # Germline variants and phasing│ ├── clair3/ # Germline SNV/indel calls│ └── hiphase/ # Phased germline variants├── somatic_snv/ # Somatic SNV/indel analysis│ ├── deepsomatic/ # Somatic variant calls│ ├── vep_annot/ # VEP annotations│ └── hiphase_somatic/ # Phased somatic variants├── somatic_sv/ # Structural variant analysis│ ├── severus/ # SV calls│ ├── svpack/ # Filtered SVs│ └── annotsv_annot/ # SV annotations├── somatic_cnv/ # Copy number variants│ └── cnvkit/ # CNVkit results├── methylation/ # Methylation analysis│ ├── pb_cpg_tools/ # CpG methylation scores│ ├── dss_dmr/ # Differential methylation regions│ └── dmr_annot/ # DMR annotations├── tumor_clonality/ # Tumor purity and ploidy│ ├── amber/ # BAF analysis│ ├── cobalt/ # Read depth ratios│ └── purple/ # Purity/ploidy estimation├── signature_analysis/ # Mutational signatures and HRD│ ├── mutationalpattern/ # Mutation signatures│ └── chord/ # HRD estimation├── pipeline_info/ # Pipeline execution reports└── multiqc/ # Aggregated QC reportFor detailed descriptions of output files, see the output documentation.
To view example results from a full-size test dataset, visit the results page on the nf-core website.
Credits
nf-core/pacsomatic was originally written by Wenchao Zhang (@wzhang42) and Haidong Yi (@haidyi).
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #pacsomatic channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.